[Notice 2013/12/30] This version is kept in case of reproducibility. We are sorry the database for CoCiter v2.4 is damaged, while we are fixing the database you can use this Version.
Welcome to CoCiter!
1. Introduction
2. Submitting queries
3. Waiting page
4. Interpreting results
5. Checking jobs
6. Web service
7. Performances
8. FAQs
If you make use of CoCiter, please cite:
Qiao N, Huang Y, Naveed H, Green CD, Han J-DJ (2013) CoCiter: An Efficient Tool to Infer Gene Function by Assessing the Significance of Literature Co-Citation. PLoS ONE 8(9): e74074. doi:10.1371/journal.pone.0074074
1. Introduction
A routine approach to inferring functions for a gene set is by using function enrichment analysis based on Gene ontology (GO) or Kyoto Encyclopedia of Genes and Genomes (KEGG) curated terms and pathways.
However, such analysis requires the existence of overlapping genes between the query gene set and those annotated by GO/KEGG.
Furthermore, GO/KEGG databases only maintain a very restricted vocabulary.
Here, we developed an algorithm called "CoCiter" (implemented in this web service) based on literature co-citations to address these disadvantages in conventional gene set and gene function analysis.
Co-citation analysis is widely used in ranking articles and predicting protein-protein interactions (PPIs).
Our algorithm can further assess the co-citation significance of a gene set with any other user defined gene sets, or to any free terms.
CoCiter is able to analyze the significance of its co-citation with two types of genes or terms:
(1) Any pre-defined/manually-curated gene sets (e.g. gene sets from GO/KEGG);
(2) Any user-defined, free term sets (e.g. "diabetes" or "leukemia").
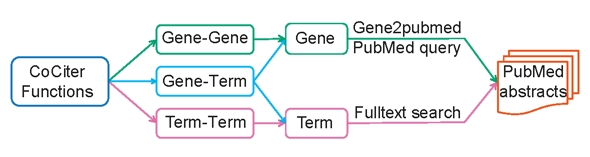
2.Submitting queries
Three kinds of queries are provided:
(1) Gene-Gene, or CoCiter Gene-Gene association analysis, calculates the significance of co-citation between two gene sets. All PubMed papers citing either gene pair from these two submitted sets would be hits.
(2) Gene-Term, or CoCiter Gene-Term association analysis, calculates the significance of co-citation between a gene set with a term set. All PubMed papers citing either gene-term pair from these two submitted sets would be hits.
(3) Term-Term, or CoCiter Term-Term co-citation analysis, provides the co-citation rate between two term sets. All PubMed papers citing either term-term pair from these two submitted sets would be hits.
They need different kinds of parameters (*: REQUIRED):
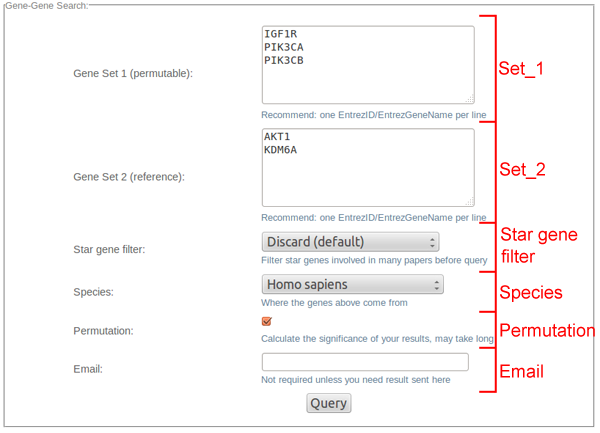
Each parameter has its own format limitations:
(1) Set_1, Set_2 (required):
-- Gene(s): a text area for line-separated Entrez gene names (case-insensitive) or Entrez gene IDs. Easy conversion from other entities with DAVID or BioMart.
-- Term(s): a text area for line-separated terms (could be phrases, case-insensitive).
(2) Star gene filter:
-- A pull-down menu to choose the filter of "star genes" (genes cited by too many papers) for the gene sets. Once filtered, they would be excluded in downstream query.
--
CoCiter by now provides two filters other than the defaulted null filter. "Filter top 0.01% cited genes" filters 822 Entrez genes in different species, while "Filter top 0.1% cited genes" filters 8154 Entrez genes in different species.
(3) Species (requied):
-- A pull-down menu to choose the species (Latin name) your genes are from. This would help a lot to interpret the Entrez gene names in the set.
--
If your interested species is not listed (or not sure which species), you could choose "Other Species(inferred)" to let the program guess. In that case, at least one Entrez ID should be given, and the first one among them would be used as a hint.
(4) Permutation:
--
A checkable box to determine whether to get the significance of co-citation by permutation.
--
For Gene-Gene queries, the permutation would take random gene sets (the same size as the first gene set submitted) from the same species to calculate the co-citation impact (CI, log-transformed paper count) with the second gene set submitted.
--
For Gene-Term queries, the permutation would take random gene sets (the same size as the gene set submitted) from the same species to calculate the CI with the term set submitted.
--
The default is 1000-times, and could only be changed to other number in the web service.
--
We currently set the seed for permutation to 123, for reproducibility.
(5) Email:
-- A text area to type in an email address for the result. Besides, we also provide job IDs to access your recent queries if you leave this parameter null.
3. Waiting page
Generally, CoCiter is quite fast to get the result. However, if a large set is involved in permutation, it might take several minutes to get the results. In that case, we provide a waiting page to automatically show the users how much calculation time is left (estimated), with a job ID and a job status page to bookmark. The page is redirected to the result page once it finished.
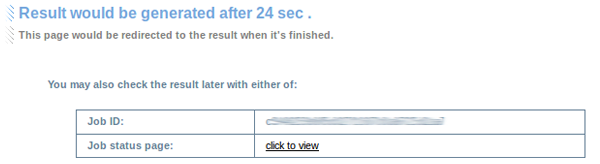
Please note that the speed has been well optimized; a 1000x1000 Gene-Gene query with 1000 permutations takes at most 4 minutes.
4.Interpreting results
Corresponding to the three kinds of queries, there are three kinds of results with similar domains.
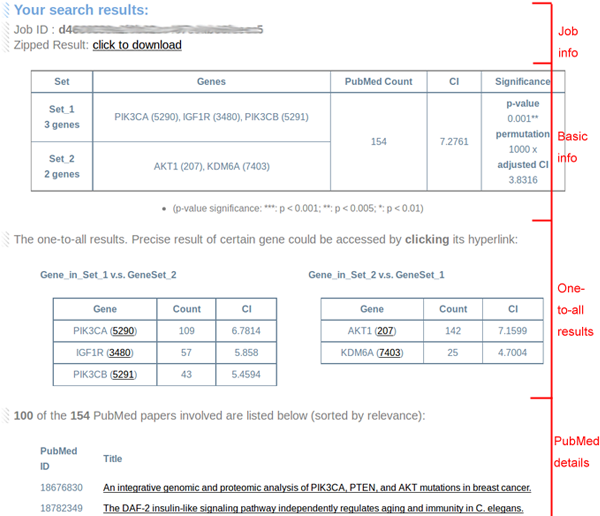
Each domain contains sub-info and exhibits part of interested results.
(1) Job info:
-- Job ID: The job ID of this result for user to check in the job status page.
-- Zipped result: The hyperlink for a dynamically generated zipped file of this result.
(2) Basic info:
-- Error (If any): The detailed report if there is some error of submission sets. Please note CoCiter would ignore input errors if there are still several genes/terms which could work.
-- Set accepted: The genes or terms used in the co-citation search.
-- PubMed count: The number of co-cited PubMed papers hits.
-- CI: Co-citation impacts, log2-transformed PubMed Count.
-- Significance: The p-value, permutation times and adjusted CI (difference between the obsearved CI and the average of permutated CI). NA for the Term-Term queries.
(3) One-to-all results:
-- To show the detail of each gene/term, their one-to-all results (one gene/term to another set) results are as well exhibited and ranked by their one-to-all CIs. Precise information could be accessed by click on their hyperlinks.
(4) PubMed details:
-- The precise list of the IDs and titles of all PubMed paper hits. The "star" or "omics" papers including too many (e.g. 100) genes are listed separately at the bottom.
The highlighted abstracts (see examples below) exhibit when hovering on the titles. The external PubMed link of interested paper could be accessed by clicking on the titles.
-- The list is sorted by highlight counts descending. If there are too many hits, we only show 100 of them. There could be several abstracts without highlights, for that gene2pubmed table includes genes showing in the text and we do not highlight genes/terms shorter than 3 characters. When there are too many genes/terms in one query (say, 5000 genes to a term), the highlight function would be discarded automatically.
5. Checking jobs
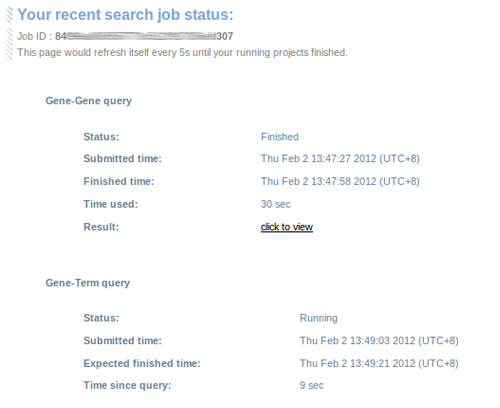
CoCiter provides job IDs and status pages for Gene-Gene and Gene-Term queries. That page, which could be accessed by URL or query the encrypted ID, would exhibit statuses of recent queries: either "No query", "Running" or "Finished", and refresh itself to show the result hyperlink.
6. Web service
CoCiter provides a Simple Object Access Protocol(SOAP) API for more sophisticated users to customize parameters and access the service programmatically.
An examplary Python script is also provided to facilitate customization (click to download).
We also provide scripts with examples (click to download) to facilitate common functional analysis.
Available Operations & WSDL:
(1) gene2gene(geneListA, geneListB, taxID, permTime, html_render)
-- geneListA, geneListB: String, '||'-delimed EntrezIDs or EntrezGeneNames.
-- taxID: Integer, taxID of the species. (e.g.: 9606 for human; 10090 for mouse; 0 for other species)
-- starGeneFilter: String, the filter name for star genes in both geneList. (default:'Discard', range:{'Discard', 'top001p', 'top01p'})
-- permTime: Integer, the number of permutation to calculate p-value; no permutation if 0. (default:1000. range: [0, 1000])
-- html_render: Boolean, if True, the result would contain a rendered html string. (default: False)
(2) gene2term(geneList, termList, taxID, permTime, html_render)
-- geneList: String, '||'-delimed EntrezIDs or EntrezGeneNames of genes.
-- termList: String, '||'-delimed terms.
-- taxID: Integer, taxID of the species. (e.g.: 9606 for human; 10090 for mouse; 0 for other species)
-- starGeneFilter: String, the filter name for star genes in the geneList. (default:'Discard', range:{'Discard', 'top001p', 'top01p'})
-- permTime: Integer, the number of permutation to calculate p-value; no permutation if 0. (default:1000. range: [0, 1000])
-- html_render: Boolean, if True, the result would contain a rendered html string. (default: False)
(3) term2term(termListA, termListB, html_render)
-- termListA, termListB: String, '||'-delimed terms.
-- html_render: Boolean, if True, the result would contain a rendered html string. (default: False)
(4) helloCoCiter()
-- Return our welcome. No input parameters needed, just for testing connection.
Please note that CoCiter Team reserves right to suspend any improper uses of the web service without notice.
7.Performances
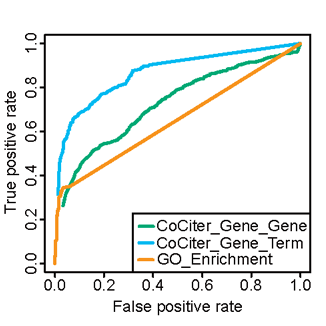
CoCiter provides an alternative and more precise approach to analyzing gene sets functions, compared with the traditional functional enrichment analysis.
Below is the receiver operator curve(ROCs) of CoCiter and GO enrichment analysis by Fisher exact test.
The analysis is based on 2097 golden standard positives (GSP) and 603 golden standard negatives (GSN) selected from the overlapping/non-overlapping annotations from GO and KEGG terms.
ROC for gene set to gene set co-citation (CoCiter_Gene_Gene) was obtained by using the KEGG genes and GO genes as input, while ROC for co-citation of gene set to a term (CoCiter Gene_Term) was obtained using the KEGG pathway keywords as terms and GO genes as input.
8.FAQs
Please feel free to contact us if anything unclear.
Q: How long would I wait for the result?
A:
First of all, we are trying to provide the best performances, both time and accuracy. However, since it's related to permutation and seraching through larget database, it might take some time when there are many or "star" genes/terms. Nonetheless, using the CoCiter job ID, you can access the results once the job finishes. Moreover, if an email address is provided in the query, the result will be sent there once the job finishes.
Q: Does CoCiter filter those "omics" papers citing too many genes?
A:
No, we would prefer keeping them.
It is true that some papers are linked to a large number of genes.However, it might not be appropriate to ignore them by simply removing them.
We have solved this problem by using the permutation approach, if one paper is linked to a large number of genes, it will be more frequently hit in the permutation process, thus will not affect the p value too much.
Assuming one paper that covers ten percent of all the genes, when random sampling a gene one thousand times, the chances that the gene is linked with the paper is 0.1, so the final p value will be around 0.1, which means the influence of the paper to the co-citation significance of the gene is not significant. So all the papers that cover more than 1% (depending on the cutoff you choose) of all genes, which can be regarded as "star" papers, will be determined as insignificantly linked to a gene this way.
To make it better understood, we show one metric in the result page named “adjusted CI”, which is defined as the difference between the observed CI and the average of permutated CIs, Meanwhile, the "star" papers including many (e.g. 100) genes are moved to the bottom of result list.
Q: Does CoCiter filter those "star" genes cited by too many papers?
A:
Not by default, but you may recognize we provide a "Star gene filter" option when you submit queries. "Filter top 0.01% cited genes" filters 822 Entrez genes in different species, while "Filter top 0.1% cited genes" filters 8154 Entrez genes in different species.
Note the cutoffs are set arbitrarily considering the amount of filtered genes.
Q: What if I have one gene list but no idea what species?
A:
Don't worry. Entrez gene IDs include the species information. We have implemented a method to infer species by them.
All you need to do is making sure the list contains at least one Entrez gene ID and choosing "Other Species (inferred)" for "Species" before the query.
Q: Why CoCiter queries only accept Entrez gene IDs and Entrez gene names? What if my gene entity is neither?
A:
CoCiter extracts gene citation mainly from the (expanded) NCBI gene2pubmed table. Hence, Entrez gene IDs and Entrez gene names would guarantee accurate reference.
However, don't panic if your genes are from other entities, such as Ensembl. We recommend specialized tools such as DAVID Gene ID Conversion Tool and BioMart for easy batch-wise gene ID conversion.
Q: Why are some of my input genes/terms filtered?
A:
For genes, those IDs not included in the selected/inferred species, and symbols different from Entrez gene names, or cited by too many papers (if certain "Star gene filter" is selected) would be filtered to guarantee the accuracy of result.
For terms, some common words like pronouns and prepositions that occur frequently in papers, which may bias the result. We filter such frequent non-scientific terms (articles, conjunctions, demonstratives, prepositions, pronouns, interjections and several verbs) adapted from a list of top 5000 frequently-used words (Jul 18, 2012 version, http://www.wordfrequency.info).
Note this filter would only be applied to terms rather than words - for example, "down" would be filtered while "knock down" would be OK. Please feel free to contact us if you find some that should not be filtered in your field.
Q: Could I use CoCiter to do functional analysis based on known data sets such as GO and KEGG?
A:
Yes. It's CoCiter's advantage that free term could work and even perform better.
If you prefer the traditional way (compare one set to many), we have provided the API and scripts with examples (click to download) to facilitate it.
Q: Could I use CoCiter to check protein-protein interactions (PPIs)?
A:
Yes. Co-citation of gene pairs has already been used as an evidence to build the PPI network (Szklarczyk, et al. 2011), and shown to support the quailities of interactions (Yu, et al. 2008). We have double checked with CoCiter, and find higher CIs are observed among high confidence PPIs.
Q: What are the databases? How often would CoCiter update them?
A:
CoCiter is based on two major datasets:
1) The extended gene2pubmed dataset that contains 38,261,321 records (downloaded from NCBI FTP in Oct 2011, expanded by NCBI E-utilities on Apr 2012);
2) The 1,640,530 abstracts from 16 common organisms (downloaded from NCBI FTP on Oct 2011).
Both datasets have very small increase compared with themselves each year. Therefore, we plan to update the datasets semi-annually.