膜蛋白在生命体中发挥着跨膜信号转导和物质转运等重要的作用。不同于可溶蛋白,膜蛋白表面有很大一部分氨基酸残基暴露于生物膜的疏水环境中。然而,膜蛋白的这一特征在现有的蛋白质结构预测研究中却鲜有考虑,之前也没有一个明确的特征量可以从蛋白序列出发予以预测表征。日前,北京大学前沿交叉学科研究院定量生物学中心、北大-清华生命科学联合中心宋晨研究组在《PLoS Computational Biology》期刊发表了题为“Membrane contact probability: An essential and predictive character for the structural and functional studies of membrane proteins”的研究论文。该研究提出了一个新的特征量——“膜接触概率”(Membrane contact probability, MCP),用于表征蛋白质序列中各个氨基酸与磷脂膜疏水部分接触的可能性,发展了一个从蛋白质序列出发预测各残基的膜接触概率的方法,并展示了该特征量在蛋白质结构和功能预测研究中的初步应用。
近年来,基于物理方法的生物分子模拟产生了大量的数据,如何将这些数据与迅速发展的深度学习技术结合起来,是计算生物学领域的一个重要问题。在此项研究中,为了定量预测膜接触概率这一特征量,宋晨研究组创新性地利用膜蛋白分子动力学模拟数据库MemProtMD [1],结合深度卷积循环神经网络,构建了一个从蛋白质序列到蛋白质残基与膜接触概率的预测模型。在训练、验证和测试过程中,模型的预测值与模拟的观测值之间的皮尔森相关系数达到了77%左右,获得了较高的预测精度。计算结果表明,对于膜蛋白而言,膜接触概率是溶剂可及性(Solvent accessibility)的必要补充,两者的结合可较完整地预测膜蛋白的外表面氨基酸(图1)。
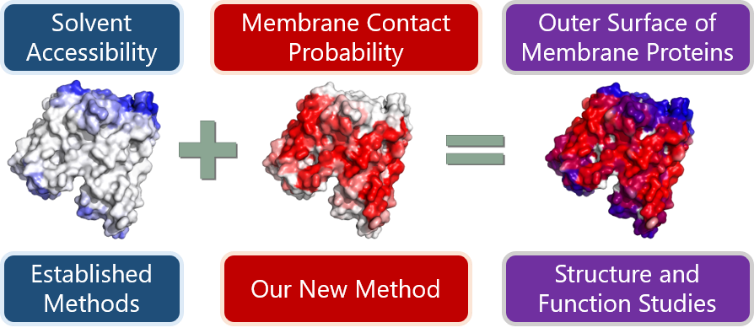
图1 蛋白质膜接触概率是溶剂可及性的必要补充。
利用该模型进行预测时,蛋白质序列为唯一输入,而预测结果会给出序列中各个氨基酸残基与磷脂膜接触的概率值。对于已知结构的膜蛋白,研究者可将预测的膜接触概率着色到相应的结构表面,会发现膜接触概率预测值高的氨基酸可形成合理的跨膜条带区(图2)。对于未知结构的膜蛋白,研究者也可预测出可能与膜直接接触的氨基酸,从而可在缺失实验解析结构的情况下对蛋白质的折叠方式和膜接触界面给出预测信息。

图2 膜接触概率在具体事例中的预测结果。膜接触概率根据数值大小以红色着色到蛋白结构表面。
此外,该研究发现蛋白质膜接触概率预测不仅对跨膜蛋白表现良好,对于膜锚定蛋白也同样适用,因此,膜接触概率的预测可以用来识别膜锚定蛋白的序列及其与膜相接触的氨基酸。在对一个可溶蛋白数据集[2]的预测中,作者筛选出了可能锚定在膜上的蛋白Sar1,并利用粗粒化和全原子分子动力学模拟进行了验证,发现膜接触概率预测和分子动力学模拟给出了一致的由N端螺旋组成的膜锚定界面(图3)。事实上,此前的实验研究结果表明,蛋白Sar1的N端螺旋很可能插入内质网膜,在囊泡运输过程中扮演重要角色[3-6],与模型预测相符。此外,在对人类蛋白组的高通量膜接触概率预测中,作者也筛选出了大量的跨膜信号肽和分泌蛋白,进一步证明了膜接触概率预测的广泛适用性,可根据蛋白序列判定其膜定位情况以及与膜接触的氨基酸残基等信息。

图3 蛋白Sar1锚定在磷脂膜表面的分子动力学(MD)模拟验证。
据此,该研究认为膜接触概率的提出很大程度上填补了膜蛋白表面结构特征的空缺,对于膜蛋白的结构预测和功能研究可以起到重要的补充作用。作为验证,该研究展示了膜接触概率在蛋白质残基接触图(Contact map)预测中的应用。2017年,许锦波教授首次将深度残差神经网络(Deep residual net, ResNet)应用到蛋白质残基接触图的预测模型中[7],极大地提升了预测精度。基于该ResNet模型,王磊等同学将预测得到的膜接触概率作为一维输入特征进行了整合,使得蛋白质残基接触图的预测精度得到了系统性的提升,部分长程接触的精度提升可达到7%左右。
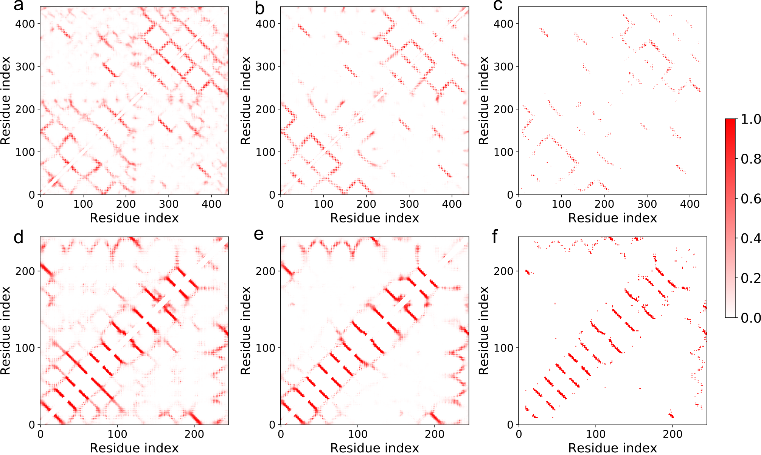
图4 在两个具体事例中蛋白质残基接触图的预测结果与相应真实结构的残基接触图。可见整合MCP信息后的接触图(b&e)比整合前(a&d)更接近真实结构接触图(c&f)。
蛋白质残基接触图预测精度的提升很可能有助于蛋白质三维结构预测,对此作者也进行了初步验证。针对两个代表性的膜蛋白结构5aym(螺旋跨膜蛋白)和4e1t(桶状跨膜蛋白),基于预测得到的蛋白质残基接触图(图4)(整合膜接触概率后top L/5预测精度分别提高10%和6%),作者利用CONFOLD2 [8]构建了可能的蛋白质结构模型。结果表明,该方法在整合膜接触概率后给出了更加精确的预测结构,符合预期(图5)。

图5 基于预测的蛋白质残基接触图构建的蛋白质三维结构。图中蛋白的真实结构用青色表示,预测结构则为灰色表示,两者的RMSD值标示在结构的左下方。可见整合MCP信息后构建的结构模型(b&d)比整合前(a&c)更加准确。
综上,该研究提出了一种全新的、重要的蛋白质特征量——膜接触概率,构建了一个基于分子动力学模拟数据和深度学习网络的预测方法,能够较准确地预测给定蛋白序列每个氨基酸的膜接触概率。该研究也表明,膜接触概率可在膜蛋白的结构和功能预测研究中发挥重要的作用。因此,宋晨研究组搭建了一个预测膜接触概率的云计算服务器(http://www.songlab.cn/),以便广泛服务于相关膜蛋白研究。需要说明的是,目前的膜接触概率预测只能表征由疏水作用驱动的一般性蛋白上膜情况,对于膜缺陷、特殊膜成分介导的上膜蛋白还无法作出准确预测,需要研究者在使用过程中予以注意。
北京大学前沿交叉学科研究院定量生物学中心博士生王磊为该论文的第一作者;北京大学生命科学学院章将国同学、定量生物学中心王大力博士和PTN项目的康凯同学参与了该研究工作并做出重要贡献。北京大学前沿交叉学科研究院定量生物学中心、北大-清华生命科学联合中心宋晨研究员为文章的通讯作者。北京大学数学科学学院邓明华教授以及美国芝加哥丰田计算技术研究所许锦波教授在讨论中给予了宝贵意见。该项工作得到了国家自然科学基金委、科技部重点研发计划和北大-清华生命科学联合中心等方面的经费资助和支持。北大-清华生命科学联合中心的“北极星”高性能计算平台为该研究提供了部分计算资源。
膜接触概率云计算服务器:
http://www.songlab.cn/
原文链接:
https://doi.org/10.1371/journal.pcbi.1009972
参考文献
1. Stansfeld PJ, Goose JE, Caffrey M, Carpenter EP, Parker JL, Newstead S, et al. MemProtMD: automated insertion of membrane protein structures into explicit lipid membranes. Structure. 2015;23(7):1350-1361.
2. Jones DT, Buchan DWA, Cozzetto D, Pontil M. PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics. 2012;28(2):184-190.
3. Huang M, Weissman JT, Beraud-Dufour S, Luan P, Wang C, Chen W, et al. Crystal structure of Sar1-GDP at 1.7 Å resolution and the role of the NH2 terminus in ER export. Journal of Cell Biology. 2001;155(6):937-948.
4. Long KR, Yamamoto Y, Baker AL, Watkins SC, Coyne CB, Conway JF, et al. Sar1 assembly regulates membrane constriction and ER export. Journal of Cell Biology. 2010;190(1):115-128.
5. Kurokawa K, Suda Y, Nakano A. Sar1 localizes at the rims of COPII-coated membranes in vivo. Journal of Cell Science. 2016;129(17):3231-3237.
6. Lee MC, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell. 2005;122(4):605-617.
7. Wang S, Sun S, Li Z, Zhang R, Xu J. Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Computational Biology. 2017;13(1):e1005324.
8. Adhikari B, Cheng J. CONFOLD2: improved contact-driven ab initio protein structure modeling. BMC Bioinformatics. 2018;19(1):22.







